FDA否决了 rociletinib(CO-1686)通过快速审批的申请3 h& [/ w- R H6 X( ?( ?% u
摘 要:近期 FDA 肿瘤药物顾问委员会小组否决了 rociletinib(CO-1686)通过快速审批的申请。小组将现存的两项 rociletinib 治疗晚期 EG
+ d; ~$ R# K) g, s V2 Q! F+ E1 l3 s* ?$ E0 p2 O. j: I
近期 FDA 肿瘤药物顾问委员会小组否决了 rociletinib(CO-1686)通过快速审批的申请。小组将现存的两项 rociletinib 治疗晚期 EGFR T790M 突变患者的两项研究的数据进行了 pooled 分析。结果总缓解率远低于该药之前所宣称的53%,同时存在更多更为严重的不良事件,包括较高的高血糖和 QT 间期延长发生率。小组认为该药应在 FDA 最后决定确定以前,提交随机 Ⅲ 期试验 TIGER-3 研究的结果。医脉通报道。7 Q" x, {6 \& d, \6 @# l" s
6 m) M" i8 G/ u7 u) u2 X" F2 t
近期,FDA 肿瘤药物顾问委员会(ODAC)以12:1的投票结果,否决了 rociletinib 用于 EGFR 靶向治疗经治的转移性 EGFR T790M 的非小细胞肺癌(NSCLC)患者治疗的快速审批。; r& y; L3 w8 f0 v. l, n
. v0 j! H; E7 w. ]3 V6 |委员会小组对早期试验 CO-1686-008(TIGER-X)研究和 CO-1686-019(TIGER-2)研究进行了 pooled 分析,考察了该药的有效性和安全性证据,是否足以支持这个三代 EGFR 抑制剂通过快速审批。除了最终的投票否决结果外,小组还建议应在 FDA 确定最终审批决策前提交随机 Ⅲ 期试验 CO-1686-020(TIGER-3)研究的结果。
; O: E( K( D: ^* o' f" j/ S! c: i; ]# `2 V2 `) a5 ?
开放标签的国际性试验 TIGER-3 研究在接受过 EGFR-TKI 和含铂两药化疗并发生进展的 EGFR 阳性 NSCLC 患者中,对 rociletinib 与单药化疗(培美曲塞、多西他赛或吉西他滨)的疗效进行了比较。3 U; M Y9 Q9 E# v- ?) {0 x- e
1 J+ P+ ~: w5 n目前的日程表显示,FDA 关于 rociletinib 的最终审批决定将于2016年6月28日确定。
# b+ k. j& h) p- ]7 I6 ^9 w, G2 \
) X- H7 i9 M1 l' O" ^2 b. g$ \! T“快速审批的要求是该方案比现有治疗更优越,但我目前并没有看到有足够的数据证明这一点”,小组的主席,Johns Hopkins 大学 Sidney Kimmel 综合癌症中心的肿瘤科教授 Deborah K. Armstrong 博士解释道。
* R$ y0 j6 h9 O1 o$ z8 L) Z" N& y, x1 f2 S/ `& S! n2 Q6 N% P
“还有太多问题没有答案,仍需解决。我同样也在担心 TIGER-3 研究能否解决所有必要的问题”,美国国家研究院(NCI)的高级研究员,药学博士 William Figg 补充道。
$ k2 m: R2 e' q& p! }2 X, [- i# ?8 J) i, b1 Y ~
这项纳入了 TIGER-X 研究和 TIGER-2 研究的 pooled 分析的结果显示,接受 rociletinib (剂量范围:500-750mg 一天两次)的325位 EGFR-T790M 转移性 NSCLC 患者的总体缓解率(ORR)为30.2%(95%CI 25.2-35.5)。接受 625mg 剂量(n=170)和 500mg 剂量(n=79)的患者,ORR 分别为32%(95%CI 25-40)和23%(95%CI 14-34),中位缓解持续时间分别为8.8个月和9.1个月。
5 [& v! d9 v+ W' m
7 m/ \5 Q+ p( A$ Y安全性方面,ODAC 小组的 pooled 分析纳入了 TIGER-X 研究和 TIGER-2 研究中的400位患者,患者接受的剂量分别为 500mg、625mg、750mg 或 1000mg 一天两次。
; \* h) n# J, ^% W# M! J
$ q/ W7 G1 \6 j2 O7 J. ]在最常见的全等级不良事件(AE)方面,发生率超过30%的 AE 有腹泻、高血糖、乏力、恶心、食欲下降,QT间期延长及呕吐。最常见3/4级 AE(超过10%)为高血糖和 QTc 延长。
( h5 q/ V# o# b8 x; t3 { f$ ~ ^ u0 z0 q
51%患者发生减药,最常见原因为高血糖(22%)和 QTc 延长(11%)。57%患者发生剂量中断,最常见原因为高血糖(22%)、QTc 延长(10%)及恶心(10%)。11%患者发生 AE 导致的停药,最常见原因为 QTc 延长(2%)及肺炎(2%)。1 ], ~0 I. C9 R. c
& y; F2 L$ H- }9 {# s47%患者发生严重 AE,最常见为恶性肿瘤进展(16%)、高血糖(8%)及肺炎(4%)。基线后测定值至少出现一次 QTc 超过500毫秒的患者有17%。1例尖端扭转型室速,2例猝死(分别发生于服药第4天和第13天)。
9 ]2 f6 s6 Z6 H7 Y2 t3 Q/ e
8 d" p; x" {1 C& j, S起初 rociletinib 的滚动申请已于2015年7月完成,随后获得了 FDA 优先审核权。然而,在事先安排的90天审核会议上,TIGER-X 研究和 TIGER-2 研究的缓解率的改变促使 FDA 要求进一步的数据。) x9 l, N$ i+ a- b# t6 I
0 Q5 g% ?" ~; d9 l, ^& H E0 S向 FDA 提交的 TIGER-X 和 TIGER-2 的早期数据显示,全部剂量水平的 T790M 突变患者(n=243)中,ORR 达到53%,疾病控制率达到 85%。% c+ n* _: D6 J, e- @( i1 i" H
3 {1 E! L7 c2 |1 j$ m, [
TIGER-X 纳入了456位 EGFR 阳性的 NSCLC 患者,接受了4种剂量(500-1000mg)。患者中位年龄63岁,10%有糖尿病史,41%有中枢神经系统(CNS)转移。之前接受的治疗中位数为2线,44%患者接受过1种以上 TKI。, `" j: b& w" s7 t; m
( v; I) w! Y( i1 a& [9 Y( g
该研究数据截止至2015年4月27日,500mg 组和625mg 组中全部 T790M 突变患者(n=270)的中位无进展生存(PFS)时间为8.0个月。基线无 CNS 转移的患者,中位 PFS 为10.3个月。目前该公司并未公布进一步的 PFS 数据。) k; v% }- {: w: u5 t! H+ o, u/ O7 Z
5 V' G* W- K6 ^% }& j* d7 s
安全性分析方面,500mg 组最常见的全等级 AE 为高血糖(35%)、腹泻(33%)、乏力(29%)、食欲下降(15%)、肌肉痉挛(14%)、体重减轻(10%),及呕吐(8%)。$ v; C$ l& u) }3 h- v, H
& {/ ]6 S6 a5 C, r
2.5%患者出现3级 QTc 延长。500mg 组未出现间质性肺炎。2.5%患者出现 AE 导致的停药。+ T2 D( s& ^8 i- a. L: K: J
; [% O, N% P* z" ^9 D500mg 组3/4级糖尿病发生率为17%。为了控制这个问题,研究人员制定了一个检测血糖并在必要时给予口服胰岛素增敏剂的方案。在该方案实施之前,3/4级高血糖发生率为22%,而后来发生率降为8%。
6 l3 Q( t3 U) W3 Y2 |% ?. A7 O, @: K, s* p9 h
正在进行的单臂 Ⅱ 期试验 TIGER-2 研究正在考察 rociletinib 二线治疗 T790M 突变的 NSCLC 患者。
; W3 q2 N# u- ^% p! x& Y& l
4 J5 |( [7 O5 f( \在该类疾病治疗领域存在已获批的药物 osimertinib(AZD9291) 也是小组中一些专家投反对票的原因。2015年11月,FDA 通过了 osimertinib 的快速审批,用于治疗前线 EGFR TKI 治疗后进展的 EGFR T790M 突变阳性 NSCLC 患者。批准依据的证据来自于两项单臂研究的411位患者数据。其中 AURA 研究的 ORR 达到61%,AURA2 研究的 ORR 达到57%。
' R8 f; e* f( w5 E7 P8 P2 M9 ^! c; L4 q9 |9 P
“一个无法回避的事实就是,osimertinib 已经通过了快速审批,并且表现出的毒性更低。因此我认为尚需更多数据”,小组成员之一,Mayo 诊所医学助理教授 Grzegorz S. Nowakowski 博士表示。
% h, {4 X- W' o q: m6 R, E6 N! I# L( C1 H
“目前已存在该领域其它方案所提供的风险-获益比。如果目前没有针对 T790M 阳性的 NSCLC 患者的药物,可能投票时我会非常纠结”,NCI 胸腔及胃肠道肿瘤科相关研究医生 Arun Rajan 博士如是说。
! \0 Q7 w" O8 m$ G2 r7 B) D
( P, }$ k, N% D- u+ c& [小组中唯一投票,认为 FDA 应继续跟进审批程序,而不需要 TIGER-3 研究结果的专家是作为小组消费者代表的 Michele Orza 博士。他解释道:“我投赞成票时也感到十分勉强。只是我认为到2018/2019年(TIGER-3 研究的预计完成时间),需要等待的时间太久了。这不代表我会投票认为该药可以通过快速审批。目前有非常多的问题需要解决,并且我也没有信心认为完成了 TIGER-3 就能够回答所有小组认为应回答问题。但同时我所考虑的是,在这段期间可能会有患者能从该药获益。我们应当努力找出这类特定的患者群,并为这类患者提供快速审批。”
2 L8 [3 n( o4 `; a" n( z
. w# M, R: y! x( i0 Z |
 汇总4月最新:临床试验招募| 小细胞
"化疗±免疫后进展了,还能怎么办?"——这是许多小细胞肺癌患者和家属的困境。
临
汇总4月最新:临床试验招募| 小细胞
"化疗±免疫后进展了,还能怎么办?"——这是许多小细胞肺癌患者和家属的困境。
临
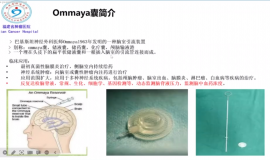
 神奇的ommaya囊——学习笔记
一个神奇的装置-ommaya囊——————9.28 苗茜医生科普讲座
直播链接:https://live.
神奇的ommaya囊——学习笔记
一个神奇的装置-ommaya囊——————9.28 苗茜医生科普讲座
直播链接:https://live.
 母亲肺腺癌晚期骨转移,EGFR21突变,
母亲1970年出生,56周岁,身高163cm,体重98kg(196斤)。2025年9月23日肺炎住院,胸
母亲肺腺癌晚期骨转移,EGFR21突变,
母亲1970年出生,56周岁,身高163cm,体重98kg(196斤)。2025年9月23日肺炎住院,胸
 临床试验招募| 铂类化疗失败后的经治
🔥 项目题目:AL8326片治疗至少接受二线治疗方案后疾病进展或复发的小细胞肺癌患者
临床试验招募| 铂类化疗失败后的经治
🔥 项目题目:AL8326片治疗至少接受二线治疗方案后疾病进展或复发的小细胞肺癌患者
 这个春天,我想和你分享!ALK+“从容
作者:pear
宠辱不惊,闲看庭前花开花落;去留无意,漫随天外云卷云舒。
这份古人笔下
这个春天,我想和你分享!ALK+“从容
作者:pear
宠辱不惊,闲看庭前花开花落;去留无意,漫随天外云卷云舒。
这份古人笔下






 显身卡
显身卡
.jpg")
.jpg")
.jpg")
.jpg")